Abstract
Clinical trial monitoring is an essential component of any clinical study, ensuring subject safety, data quality, and protocol compliance. In recent years, industry experts and regulatory agencies like the US Food and Drug Administration (FDA) and the International Conference of Harmonization (ICH) have advocated a risk-based approach to clinical trial monitoring.
There is an increased emphasis on critical data and processes, with greater use of remote, centralized monitoring rather than on-site monitoring. This approach allows sponsors and Clinical Research Organizations (CROs) to help detect and manage clinical trial risks earlier rather than later.
The global pandemic, the adoption of Decentralized Clinical Trials (DCT), and the amplified role of information technology has catalyzed the wide-scale acceptance of Risk-Based Monitoring (RBM).
What is Risk-Based Monitoring in Clinical Trials?
The Covid-19 pandemic has changed the way clinical research is conducted. It has been a catalyst for moving away from inefficient practices and standardizing new clinical trial operational models to ensure faster completion of clinical studies. To complement this, as remote and virtual site monitoring approaches become mainstream, many organizations have been forced to adopt risk-based approaches. This, in turn, has further accelerated initiatives by the FDA, which included ICH E6 (R2) as a revision to the Good Clinical Practice (GCP) in 2016 and required sponsors to develop risk-based methods for assessing and managing trial risks.
The pandemic further proved that the drug development industry can no longer rely on inefficient trial monitoring models and must instead implement processes prioritizing data-driven predictive risk assessment and mitigation. The traditional approach of on-site monitoring of clinical trial sites has helped ensure that studies reliably assess the safety and efficacy of experimental treatments. This has been achieved through source-data verification (SDV) or by comparing the data collected in the case of report forms (Case Report Forms) to those in the patient’s medical records. However, it is a very time-consuming process, taking more than half the time of site monitoring visits, and imposing a high work burden on onsite personnel.
Risk-based monitoring applies the principles of Quality by Design (QbD) and risk-based management to all clinical study elements, from planning to execution. It starts with critical processes and data identification. These identified critical processes and data are monitored remotely throughout the trial life cycle. By using predictive analytics models, it is possible to identify and evaluate risks. This helps ensure that instead of reactive corrective and preventive actions, a proactive approach encompassing risk control, risk communication, risk review, and risk reporting can be maintained, beginning from the first site initiation till the last site closure.
Leveraging information technology is not a choice but the one and only option for successfully implementing risk-based monitoring in clinical trials. Predictive online analytical processing is essential for identifying and evaluating high-risk sites and data points, and web-based interfaces are required for remote and virtual monitoring of such high-risk data.
A holistic risk-based quality management system is essential for clinical study monitoring. This system, involving both centralized planning and decentralized monitoring of risks is directed by the risk planning team and makes efficient use of information technology.
Designing a Risk-based Monitoring Management System
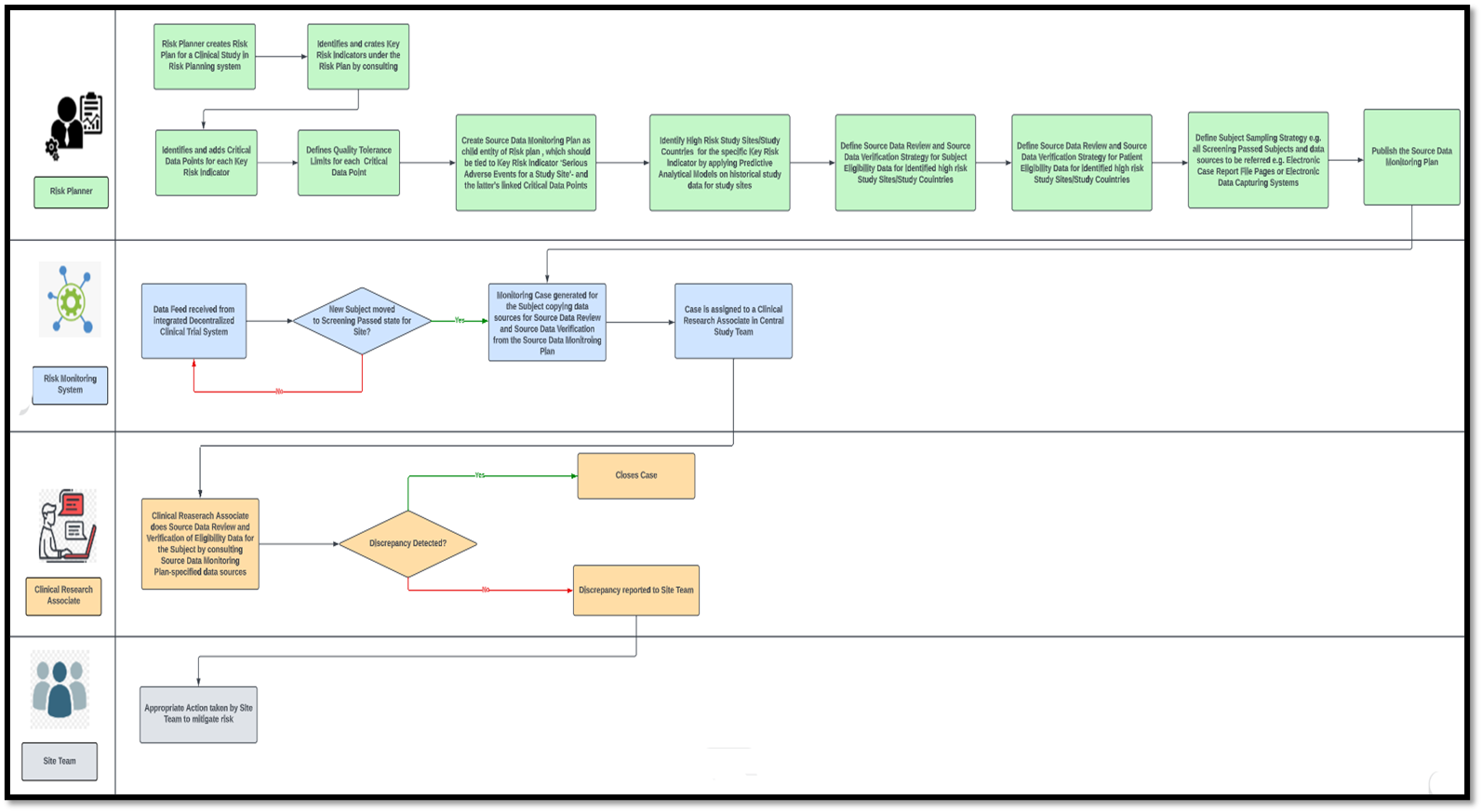
Any risk-based clinical study monitoring system will typically comprise two major modules— a risk planning module and a risk monitoring module. A risk plan is the topmost entity in the hierarchy of entities used in a risk planning system. The sponsor’s clinical study design team uses the risk planning module.
One risk plan can be reused for multiple clinical studies with similar designs and endpoints. Within a study’s risk plan, Key Risk Indicators (KRI) are defined. A KRI may be at various levels—study, study country, study site, or subject level. For example, ‘Serious Adverse Events for a Study Site’ may be a site-level KRI. Multiple Critical Data Points (CDP) must be tied to KRIs like ‘No. of Site-Level Serious Adverse Events’, ‘Ratio of Serious Adverse Events to Number of Subjects for the Site’ etc. A study-level Quality Tolerance Limit (QTL) must be set for each CDP.
While the risk plan outlines the parameters essential for measuring risk in clinical trial-based monitoring, executing these measurements and strategies for risk mitigation are articulated within the operational plans. Several operational plans can be defined for a single risk plan, each dictating the risk monitoring and mitigation methodologies for a single KRI or combination of KRIs.
The source data monitoring plan is an example of a commonly used operational plan that can measure and mitigate different KRIs. In conventional clinical studies, source data monitoring had to be done at the site during various stages of the site life cycle by site monitors. The site monitor had to go through the source data collected for each subject at the site, captured either on paper or on an Electronic Data Capturing (EDC) system. This was necessary, as even if for one out of a hundred subjects incorrect data is captured (eligibility data, consent data, or effect of investigational drug administration data) that violates the study protocol, it would be dangerous for subject safety and jeopardize the entire study, leading to the loss of time and resources for the sponsor. Nevertheless, such conventional monitoring is cumbersome, time-consuming, highly inefficient, and incompatible with the post-pandemic world of decentralized clinical trials.
In the risk-based monitoring ecosystem, a source data monitoring plan would be a child entity of the study’s risk plan. A KRI like ‘Serious Adverse Events for a Study Site’ can be linked to the Source Data Monitoring (SDM) Plan, along with the KRI’s CDPs. The structure of the SDM plan will be determined by predictive analytical models applied to historical study data. These predictive analytics will pinpoint not only high-risk study sites but also high-risk study countries for global clinical trials, discerning sites, and countries that are more prone to serious adverse events.
The source data monitoring plan establishes the methods for blinding and unblinding essentials for Source Data Review (SDR). Comparing the recorded data against the source documents ensures a match as per the plan. Additionally, if necessary, it will define Source Data Verification (SDV), ensuring the quality of source documentation for subjects enrolled in the study within high-risk countries or sites. The plan will also encompass the determination of datasets to undergo Source Data Review (SDR) or Source Data Verification (SDV), accounting for potential variations across distinct study sites and events. For example, an SDM plan may specify that eligibility data must be SDR-ed and SDV-ed for each subject who passes screening for Site A. For Site B, it could be the laboratory data for a subject that has to be SDR-ed and SDV-ed after each investigational drug administration visit. For Site X, only SDR may be required for the same data, but for a higher-risk Site Y, both SDR and SDV might be the strategy defined in the SDM Plan.
How will source data review and source data verification be done based on this SDM plan?
This is where the risk-based monitoring system comes into play. Imagine that the SDM plan states that for Site M, eligibility criteria matching must be checked for each subject who passes screening. When a subject for Site M in the decentralized clinical trial management system (integrated with the risk-based monitoring system) is marked as "Screen-Passed," an automatic case is created for that subject in the risk-based monitoring system. The instructions for blinding and unblinding, along with the eCRF pages’ housing subject screening data and the Electronic Data Capturing (EDC) system for data entry, seamlessly transfer from the SDM plan to the case.
Furthermore, an automatic assignment of the case to a Clinical Research Associate (CRA) or monitor will occur. The CRA will run an online review and verify source data from the eCRF and EDC systems, respectively. The case will be closed if no discrepancy is found. If any discrepancy is found, the CRA will immediately report it to the site, and the site team must take appropriate action.
Cases may not be generated for every screen-passed subject; rather, they will be created for every nth set of five subjects transitioning to the screen-passed state at the site. Alternatively, cases might also be established exclusively for subjects aged 45 years and above, as outlined in the sampling strategy detailed in the SDM Plan.
Sample Process Flow Diagram of a Risk-Based Monitoring System for Source Data Monitoring of Subject Eligibility
Conclusion
As decentralized clinical trials become the norm rather than the exception in the changing landscape of drug development, risk-based monitoring will evolve from a choice to the most feasible option. As technology continues to merge with medical science, exciting possibilities are on the horizon.
The near future promises the emergence of groundbreaking therapies like gene therapy, seamless EHR interoperability, revolutionary advancements in electronic patient-reported outcome technology, and the integration of highly advanced AI-ML algorithms for enhanced predictive analytics. Considering these developments, the landscape of risk-based monitoring in clinical trials is poised for remarkable transformation, diverging significantly from its current state.
Acknowledgment
This piece was written by Arup Ghosh at Encora.
About Encora
Fast-growing tech companies partner with Encora to outsource product development and drive growth. Contact us to learn more about our software engineering capabilities.

